Reviews
Vol. 45: Issue 3 (Suppl. 1) - June 2025
Exploring the genetic landscape of otosclerosis: current understanding and future perspectives
Summary
Cover Image
Otosclerosis is characterised by abnormal bone remodelling in the otic capsule, leading to progressive hearing loss. Unlike many genetic disorders, the causative genes for otosclerosis remain largely unidentified despite extensive research using linkage analysis and genome-wide association studies (GWAS). Inheritance patterns in otosclerosis suggest a multifactorial model involving genetic predisposition and environmental triggers, a model applied to other common diseases, such as age-related hearing loss, coronary artery disease, and Alzheimer’s disease. Linkage analysis has identified nine loci associated with monogenic forms of otosclerosis, yet the specific causative genes and variants remain elusive. Promising insights have emerged from GWAS, with strong associations identified for novel candidate regions, including the RELN gene. Recent studies using next generation sequencing have identified several candidate genes such as SERPINF1, ACAN, and MEPE. SERPINF1, encoding pigment epithelium-derived growth factor, is linked to regulation of angiogenesis in bone remodelling. ACAN, associated with the OTSC1 locus, encodes aggrecan a crucial component of the extracellular matrix in cartilage, showing a range of variants with varied effect sizes and frequencies. MEPE, involved in bone homeostasis, has been significantly associated with otosclerosis in large family-based and casecontrol cohorts. While considerable progress has been made in identifying potential genetic contributors, the precise genetic architecture of otosclerosis remains to be fully elucidated. An integrated approach combining genetic data and clinical information, such as audiometric testing and temporal bone imaging, is essential for a comprehensive understanding of otosclerosis.
Introduction
Otosclerosis is a disease of bone remodelling that results in localised bone dysplasia within the otic capsule, causing progressive conductive hearing loss in 90% of affected patients, with a sensorineural or mixed component in the remaining 10% 1. Reports of hereditary conductive hearing loss, consistent with clinical otosclerosis, date back to the late 19th century 2. Subsequent studies have identified patterns of inheritance, including autosomal dominant with incomplete penetrance (about 25-40%) 3 and a digenic pattern 4. Nevertheless, families exhibiting a clear Mendelian-like autosomal dominant inheritance of otosclerosis are uncommon, as the majority of cases of otosclerosis with a positive family history (50-60%) do not adhere to clear Mendelian patterns, while the remainder (40-50%) are sporadic with no family history of the disease 5.
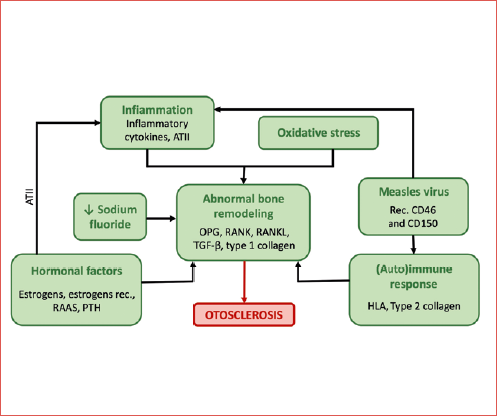
Among the numerous models to explain the inheritance patterns of otosclerosis, the most likely explanation lies in complex (multifactorial) inheritance, entailing a combination of genetic susceptibility genes and environmental factors, and their interactions, a model proposed for many other diseases, such as age-related hearing loss, Alzheimer’s, and coronary artery disease 1. However, monogenic and complex inheritance models should not be considered as separate entities, but as part of a continuum spectrum where the different genetic variants can be ordered, ranging from very rare ones with a large size effect and an almost perfect Mendelian segregation, to common variants with low effect sizes that act as susceptibility factors in a more complex and multifactorial aetiological pattern. In this model, genetic susceptibility factors interact with environmental triggers to generate the pathological phenotype. Among the possible environmental risk factors for the development of otosclerosis, infection with measles virus, low sodium fluoride in drinking water, and endocrine factors are under investigation (Cover figure).
Epidemiologically, the incidence of otosclerosis is higher in Caucasian patients of European descent, showing a prevalence of 0.2-1% in the general population 5, whereas it is rare among Africans, Asians, and American Indians 6. This disparity may reflect differences in genetic contributions and environmental risk exposures. Additionally, otosclerosis is more prevalent in females than males, at a ratio of about 2:1. This observation suggests a potential role for sex hormones in the development of pathologic otic capsule dysplasia.
Bone remodelling is a fundamental biological process that is essential for repairing bone damage, preventing the accumulation of aged bone – which may lose its flexibility and become brittle – and creating a reservoir for calcium and phosphorus. The efficacy of bone remodelling lies in the dynamic equilibrium between bone resorption and deposition. This balance is achieved through the spatially coordinated, integrated, and sequential actions of osteoclasts and osteoblasts, which collectively form a basic multicellular unit. This process is tightly regulated by various factors, including osteoprotegerin (OPG), receptor activator of nuclear factor-kappa B (RANK), and its ligand (RANKL) 7.
Mesenchymal-derived bone-forming osteoblasts express and secrete RANKL in response to a variety of hormones, cytokines, and mechanical stimuli. Upon secretion, RANKL binds to its receptor RANK on monocyte progenitor stem cells (MPC), initiating their differentiation into mature, active osteoclasts. These osteoclasts, which fuse into multi-nucleated cells, are responsible for bone resorption by secreting lysosomal enzymes, such as collagenases, and hydrochloric acid, capable of dissolving hydroxyapatite, one of the main acellular components of bone tissue. The activation and differentiation of osteoclasts through RANKL are neutralised by the decoy receptor OPG, also secreted by osteoblasts 8. Conversely, bone repair is initiated by osteoblasts that secrete the osteoid seam in the lacunae created by osteoclasts. Calcium and phosphate ions then deposit in the form of hydroxyapatite, effectively trapping the secreting osteoclasts, which differentiate into osteocytes. An imbalance of the OPG/RANKL ratio has been implicated in various bone disorders, such as osteoporosis, rheumatoid arthritis, and bone metastases 9, while inactivating mutations in the OPG gene have been demonstrated to cause juvenile Paget’s disease of bone 10.
The unique biology of otic capsule
The dense tissue of the petrous temporal bone that surrounds the membranous labyrinth of the inner ear is known as the osseous labyrinth or otic capsule. It has long been recognised as a histologically unique bone, characterised by the highest bony density in the body 11, and a rate of growth, modelling, and remodelling that is minimal compared to other bones, and virtually absent close to inner ear spaces 12-14. The primary foetal bone in the otic capsule is compact, highly mineralised 15, and formed by endochondral ossification, a process that involves a cartilaginous precursor which is resorbed and replaced by dense lamellar bone. Bone turnover rates in the adult temporal bone increase centrifugally from 0.1%/year in the innermost perilabyrinthine zone – where epifluorescence demonstrated a persistence of early foetal bone around the inner ear spaces in a rabbit animal model 16 – to about 10%/year at the capsule periphery, a rate comparable to other bones in the body 13,16,17. These observations suggest the existence of a local inner ear mechanism that inhibits perilabyrinthine bone resorption and remodelling.
In 2010, Stankovic and colleagues 18 used real-time quantitative polymerase chain reaction (RT-PCR) and in-situ hybridisation to compare gene expression between the healthy adult murine otic capsule and other bones in the body, particularly the tibia (formed by endochondral ossification like the otic capsule) and the parietal bones (formed directly by intramembranous ossification without an intermediate tissue). They found that the molecular profile in the otic capsule significantly differs from the other bones, involving a reduction of pro-inflammatory cytokines, an increased expression of anti-inflammatory cytokines, and a distinct pattern of expression of bone morphogenic proteins (BMP). The key predictors of otic capsule bone were OPG, bone morphogenic protein receptor 1B (BMPR1B), and bone morphogenic protein 3 (BMP3).
The increased expression of OPG is the most characteristic marker of the otic capsule, as established in numerous studies 19-23, and is expressed 1600 times more in the spiral ligament and 800 times more in the fluid-filled inner ear space than in other bones in the body 19 and associated with a marked inhibition of bone remodelling within the otic capsule. OPG is expressed at high levels by cochlear neurons of the inner ear and then diffuses into the surrounding otic capsule, being a central regulator not only of bone resorption inhibition but also of neurite growth stimulation. A possible explanation for the central role of the OPG-RANK-RANKL pathway in the dynamic interaction between the skeletal and nervous systems is provided by the hypothesis that RANKL may activate the expression of neurite growth inhibitor A (NOGO-A), which, when expressed, has been associated with a dramatic shortening of spiral ganglion nuclei neurites 23,24. Therefore, the competitive inhibition of RANKL by OPG has the potential to inhibit bone remodelling and favour nerve growth, aiming to prevent nerve compression by bone growth in the otic capsule. Conversely, during the early postnatal development of the inner ear, bone remodelling is active in otic capsule development and nerve growth is inhibited 23. OPG knockout mice (OPG-/-) demonstrated a degeneration of the cochlear nerve with progressive sensorineural hearing loss (SNHL) that was superimposed on a form of earlier conductive hearing loss due to resorption of ossicles in the middle ear 19,21,25. The mechanism of apoptosis caused by the loss of OPG in spiral ganglion neurons likely involves the ERK signalling pathway, an important regulator of myelination that makes neuron cells more sensitive to oxidative stress 25. In OPG-/-, this process can be rescued with medical therapies involving the administration of exogenous OPG, ERK inhibitors, or bisphosphonates 25. Alterations in the expression of OPG have been associated with diseases such as osteopetrosis 26, osteoporosis 27, otosclerosis 19,28, juvenile and adult Paget’s disease of bone 29, and celiac disease characterised by high-turnover osteoporosis 30.
BMPs play an essential role in skeleton development and repair, and have been reported to be important for otic capsule formation and maintenance of the membranous labyrinth 31,32. Conversely, inhibition of BMP by noggin has been associated with a loss of the otic capsule and membranous epithelium in an avian model 32. BMPR1B is histologically unique to the otic bone, characterised by endochondral formation, while it is absent in parietal bones, intramembranous bones whose development does not involve a cartilage intermediate 18. A lower expression of BMP3 is characteristic of the otic capsule compared to other bones 18. It is the most abundant BMP in adult trabecular bone, being a negative regulator of bone density 33; considering that the otic capsule is the densest bone in the body, the down-regulation of BMP3 is coupled with the reported increase in bone density. However, when comparing BMP3 knockout mice 33 with BMP3 over-expressor mice 34, no gross abnormalities within the otic capsule or the cochlear membranous labyrinth were reported, concluding that BMP3 has a minimal role in controlling bone remodelling in the otic capsule 18.
The downregulation of inflammation is a key molecular feature of the otic capsule, and is possibly important in the maintenance of normal hearing. Proinflammatory cytokines in the cochlea are generally considered markers of disease and have been associated with hearing loss, with anatomical communication between the otic capsule and the cochlea perilymph 19, and with pathological new bone formation within the membranous labyrinth (labyrinthitis ossificans) 35. Consistently, when comparing the healthy adult murine otic capsule and other bones in the body, Stankovic and colleagues 36 reported a lower expression of pro-inflammatory cytokines (TNFα, IL1α, IL1β, IL6, NFKβ1) and an increased expression of anti-inflammatory cytokines (particularly IL11) in the otic capsule.
Monogenic and familial forms of otosclerosis
Gene identification in otosclerosis depends on the mode of inheritance, distinguishing between confirmed familial (possibly monogenic) cases and those with a complex (likely multifactorial) inheritance pattern. For large families with numerous members affected, gene identification relies on linkage analysis to pinpoint the disease-causing variant by highlighting the chromosomal region shared by all affected individuals within a family. The effectiveness of this method hinges on the size and structure of the family selected for study. Typically, hundreds to thousands of genetic markers, which segregate in a Mendelian fashion, are analysed in a chromosomal region common to all affected family members, effectively acting as a monogene. Once the region of interest is identified, it is possible to refine the candidate loci using additional markers or genetic databases, and suspected genes can be examined through mutation analysis. Although this approach has identified nine different loci (Tab. I), pinpointing causative genes has been challenging, with only two candidate genes: T-cell receptor beta (TRB locus) identified in the OTSC 2 region on chromosome 7 36 and FOXL1 recently identified on chromosome 16 in OTSC 11 locus 37. However, even in this instance, the variants responsible for the disease remain unidentified.
OTSC 1
The first locus associated with familial otosclerosis was reported by Tomek and colleagues in 1998 38. In their analysis, they explored the impact of age on disease progression, noting that the sensorineural component of HL worsened in older subjects, whereas conductive HL did not significantly differ between younger and older subjects. Further genetic linkage analysis using short tandem repeat polymorphisms (STRPs) yielded a maximum multipoint logarithm of odds (Lod) score of 3.4 38. The Lod score, a statistical method used to assess linkage, evaluates the likelihood of a given sequence of genetic events occurring if the loci are linked as compared to its occurring if the loci are not linked. Linkage is considered to be established if the Lod score exceeds 3. Subsequent genetic analyses restricted the linked region to a 14.5 centimorgan (cM) segment between the far and near centriole segments of the long arm of chromosome 15 (D15S657). This region contains the gene for aggrecan (ACAN), the primary non-collagenous component of the cartilaginous extracellular matrix 39,40.
OTSC 2
The OTSC 2 region on chromosome 7 harbours some known genes, such as TIF1a (transcription intermediary factor 1-alpha), and PLOD3 (procollagen-lysine, 2-oxyglutarate, 5-dioxygenase 3)41. TIF1 is a growth suppressor required for the activity of retinoic acid, which has been shown to disrupt the development of the otic capsule 42. PLOD3 takes part in the biosynthesis of collagen, and in vitro expression studies have shown that PLOD3 hydroxylates lysyl residues in collagen sequences in non-triple-helical conformation. Moreover, PLOD3 activity is enhanced by tumour necrosis factor-alpha (TNFα), which is a key mediator in the pathogenesis of arthritis, causing cartilage degradation and joint destruction 43. Moreover, the region harbours T-cell receptor beta (TRB locus), which is one of the candidate genes for which evidence of association with otosclerosis was provided by Schrauwen and colleagues 1,36, describing a lower mRNA expression of TCR-beta and a decreased percentage of circulating TCR-alpha/beta-positive T cells in patients with otosclerosis linked to OTSC2 compared to controls and patients with a complex form of the disease. Further analysis showed significant disturbances in specific T-cell subsets, including an increased population of CD28null T cells in OTSC2 patients, which are considered senescent cells and whose higher proportion may indicate an altered T cell development or aging in OTSC2 patients. Overall, these findings may contribute to elucidate a possible immunological contribution to the development of otosclerosis. However, the pathological TCR-beta variant responsible for this phenotype could not be identified.
OTSC 3
The region 6p21.3-22.3 on chromosome 6 (identified as OTSC 3) includes candidate genes such as RING1 and COL11A2 44. Overall, these genes may provide insights into the association of otosclerosis with collagen abnormalities.
RING 1 together with Yin Yang 1 binding protein (RYBP) interacts with Yin Yang 1 (YY1) 45, a transcription factor activator of the COL1A1 (collagen type 1) promoter in fibroblasts 46. An abnormal COL1A1 transcription may impact the normal stoichiometry of COL1A1 and COL1A2 in the production of collagen trimers, as exemplified by osteogenesis imperfecta, a disease also characterised by fixation of the stapes footplate.
COL11A2 is a putative collagen-modulating element gene expressed in the otic capsule that causes autosomic dominant non-syndromic HL at the DFNA13 locus 47. Moreover, the region contains human leukocyte antigens (HLA) genes, consistent with the reports of a significant association of certain HLA-A and HLA-B antigens with otosclerosis 48.
OTSC 4
The OTSC 4 region on chromosome 1649 involves several genes related to bone homeostasis or immune development, including members of the cadherin superfamily (transmembrane proteins that mediate cell recognition and cell-cell adhesion), of the conserved oligomeric Golgi (COG) multiprotein complexes, involved in intracellular membrane trafficking and expressed in the immune system, and of members of the DEAD (Asp-Glu-Ala-Asp) box proteins, involved in RNA transcription, translation, export and turnover, and ribosome and spliceosome assembly 49.
OTSC 5
The OTSC 5 region on chromosome 3 involves two candidate genes: procollagen COOH-terminal proteinase enhancer protein 2 (PCOLCE2) and carbohydrate sulfotransferase 2 (CHST2). The PCOLCE2 gene product is a glycoprotein that binds the COOH-terminal propeptide of type I procollagen and is highly expressed in non-ossified cartilage in developing tissues. The CHST2 gene product is a Golgi-associated sulphotransferase, with a possible role in intercellular communication. However, mutation analysis of the coding region and the intron-exon boundaries of both genes did not reveal any disease-causing mutation 50.
OTSC 7
The candidate gene in the OTSC 7 region in chromosome 6 is represented by COL12A1 (collagen type II alpha 1) which belongs to the fibril-associated collagens with discontinuous triple helices, and is expressed in the cochlea, while further mutation analyses failed to reveal any disease-causing mutation 51.
OTSC 8
Among the genes in the OTSC 8 region on chromosome 9, Bel Hadj Ali and colleagues 52 described three possible candidates: tight junction protein 2 (TJP2), transient receptor potential cation channel, subfamily M, member 3 (TRPM3), and kruppel like factor 9 (KLF9). TJP2 belongs to the family of membrane-associated guanylate kinase (MAGUK) homologues, which take part in epithelial and endothelial intracellular junctions. TRPM3 is a cation-selective channel important for cellular calcium signalling and homeostasis and for osteoclast activity. KLF9 is a strong activator of activating enhancer binding protein 2 alpha (AP-2), which is a fundamental regulator of the mammalian craniofacial development.
OTSC 10
The region on chromosome 1 identified by Schrauwen and colleagues 53 and named OTSC10 contains 306 gene predictions, including two candidate genes: transforming growth factor beta 2 (TGFB2) and angiotensinogen (AGT), selected due to their known role in bone remodelling and on the basis of previously found associations with otosclerosis 51,55.
OTSC 11
In a recent study, Abdelfatah and colleagues 37 identified on chromosome 16 a novel OTSC locus (OTSC 11) in a Caucasian family of English extraction with a form of autosomal dominant otosclerosis who had previously tested negative for shared OTSC loci haplotypes and susceptibility genes (COL1A1, COL1A2, NOG), and for rare variants associated to the SERPINF1 gene. Sanger sequencing for 12 positional candidate genes identified an in-frame deletion in FOXL1 (NM_005250.3: c.976_990del) associated with the phenotype in the affected family, resulting in a significant loss of the protein’s helical structure. FOX proteins are a superfamily of transcription factors with a wide range of functions at the junction of multiple signalling pathways, with crucial roles in regulating gene expression in cell metabolism, proliferation, differentiation, and apoptosis 56.
Unsuccessful sequencing of functional candidate genes
To identify candidate genes of monogenic forms of otosclerosis, direct sequencing of positional candidate genes has been applied. The selected genes were prioritised based on phenotypic similarities between otosclerosis and related diseases. For example, NOG mutations cause several syndromes which share the presence of stapes ankylosis 57, while COL1A1 and COL1A2 encode alpha chains of collagen type 1, and when mutated cause osteogenesis imperfecta 58, which is characterised by a form of HL resembling otosclerosis (progressive, developing from the second-third decade of life, often both conductive and sensorineural). However, despite the resemblance with otosclerosis, this approach did not identify any disease-causing mutation 58.
Despite the 20-year lapse since the mapping of OTSC1, the OTSC genes remain refractory to discovery due to the rarity of monogenic families, diagnostic challenges, and reduced penetrance. However, the application of new approaches (such as positional cloning and next generation sequencing, NGS) as applied in the recent work by Abdelfatah et al. 37. on OTSC 11, may produce successful results in identifying all OTSC genes, which remains a fundamental step in clarifying the genetic landscape of otosclerosis.
Complex forms of otosclerosis
Although otosclerosis appears to follow a Mendelian-like autosomal dominant pattern in some isolated families, most hereditary forms of the disease result from a complex transmission. In these cases, no single genetic susceptibility factor is either necessary or sufficient to develop the phenotype; rather, it is the combination of all factors that is crucial. The typical research method to identify genetic variants of complex diseases involves candidate gene-based association studies using a case-control design. This approach, which identifies variants of selected genes that are significantly more frequent in patients than in matched controls, has been performed in numerous studies. Some genes have been found to be significantly associated with otosclerosis in more than one study, while associations with other candidate genes have not been replicated. However, lack of replicability does not necessarily rule out an association, as the sample size might have been inadequate, or different disease-causing variants may be present in different populations. The most recent alternatives to candidate gene-based association studies are genome wide association studies (GWAS) and microarray gene expression studies, which compare gene expression in diseased tissues to that in controls. With the latter method it was possible to identify different pathways to which otosclerosis susceptibility factors seem to belong, including bone remodelling and immunological, inflammatory, and endocrine pathways.
Altered bone metabolism
Collagens
The first study to hypothesise a common genetic basis between otosclerosis and osteogenesis imperfecta type 1 (caused by mutations in type 1 collagen) was published in 1998 by McKenna and colleagues 59. They provided a rationale for this association based on the similarities in histopathology and HL features between the two diseases. This initial case-control genetic association study demonstrated a significant association of clinical otosclerosis with mutations of COL1A1 in a small population of European descendants in Massachusetts. These findings were later confirmed by the same group 60, which further identified an association with otosclerosis for polymorphisms in the first intron of the Sp1 binding site of COL1A1, a finding also recently reported by Zhang and colleagues 61. In 2007, Chen et al. 62 confirmed these results and reported an association with polymorphisms that alter the binding of transcription factors regulating COL1A2, leading to an increase in COL1A1 homotrimers and a subsequent abnormal bone deposition in the otic capsule. Normally, collagen type 1 triple helices are composed of COL1A1 and COL1A2 in a 2:1 ratio, whereas COL1A1 homotrimers are rare. Associations of otosclerosis with COL1A1 have been reported in numerous studies 63,64, but not in others 65,66, and, more relevantly, were not reported in a meta-analysis of GWAS studies of otosclerosis in three population-based biobanks comprising 3,504 cases and 861,198 controls 67. Instead, this meta-analysis reported a significant association with a subunit of collagen type 4 (COL4A2), a collagen form located in the basement membrane and highly conserved across species. Mutations in other subunits of collagen type 4 have been linked to Alport syndrome, which is characterised by progressive SNHL, nephritis, and histologically by an abnormal basement membrane and dysmorphogenesis of the organ of Corti. Future studies will need to determine the role of COL4A2 as a structural or signalling element in the context of otosclerosis.
Transforming growth factor-beta (TGF-β) superfamily
The TGF-β superfamily is composed by cytokines playing a crucial role in embryonic development and maintenance of the otic capsule 68, as demonstrated by its influence on the expression of glycosaminoglycans, fibronectin, and collagen in the extracellular matrix 69. The most relevant member of the TGF-β superfamily in the context of otosclerosis is TGFβ1, a major osteogenic cytokine involved in regulating bone mass and bone matrix. TGFβ1 is expressed in the otosclerotic foci and the hyalinised spiral ligament 70. It induces several processes in connective tissues, including the promotion of collagen type 1 and fibronectin formation 71, interference with potassium circulation by affecting fibrocytes in the spiral ligament 72, induction of chondrogenesis in the otic capsule mesenchyme, and the promotion of otic capsule growth during early stages of inner ear development 73. A large association study 54 reported the Thr263Ile substitution to be significantly more expressed among otosclerosis patients than controls both in a Belgian-Dutch and in an independent French sample, and these results were replicated also in Tunisian 74, Hungarian 66 and British studies 67, but not in a black South African population 75. Additionally, sequencing the exons and intron-exon boundaries of TGFβ1 in 755 patients with otosclerosis and 877 controls revealed three rare nonsynonymous variants in four patients 76, which were not present in the controls (c.G86A, p.Gly29Glu; c.G86C, p.Gly29Ala; c.C722T, p.Thr241Ile). An analysis in an Indian sample linked the c.-509C > T single nucleotide polymorphisms (SNP) with otosclerosis, as well as a specific G-T-T-G haplotype constructed from four SNPs 77. A de novo mutation in the promoter region was also discovered in one patient, leading to decreased expression of TGFβ1 77. A study on proteomic analysis and immunostaining of temporal bones, conducted by Richard and colleagues 78, identified TGFβ1 as being expressed in patients affected by cochlear otosclerosis and hyalinisation of the spiral ligament.
The mechanism by which TGFβ1 contributes to the pathogenesis of otosclerosis remains unclear. However, one theory suggests that TGFβ1 may influence the globuli interossei within the otic capsule (residual cartilage rests within the dense bone of the otic capsule, a unique feature of the inner ear), potentially targeting these structures for an immune reaction that results in otosclerosis 78.
A meta-analysis of GWAS studies involving 3,504 cases and 861,198 controls 67 confirmed the association between TGFβ1 and otosclerosis, identifying the intronic variant rs8105161 as the strongest. Additionally, the analysis highlighted the potential roles of other genes in the TGFβ1 signalling pathway. These include RUNX2, a transcription factor essential for osteoblast and chondrocyte differentiation, regulated by TGFβ1 79. RUNX2 was found to be expressed only during the development of the otic capsule and not in its mature state. This is a critical finding, as otosclerosis may be triggered by globuli interossei, embryonic remnants within the otic capsule. Other significant genes are SMAD3 (a downstream transcription factor), CD109 (a TGFβ1 co-receptor acting as a negative regulator of the TGFβ1 pathway), LTBP3, which regulates the latency and activation of TGFβ1 through direct extracellular binding, and AHSG, which antagonises TGFβ1 signalling and directly affects the mineralisation process by inhibiting calcium phosphate precipitation. Mutations in AHSG can cause multiple-synostosis syndrome, which is characterised by stapes fixation, an otologic presentation that mimics otosclerosis 57. In a microarray analysis of otosclerotic stapedial footplates and controls, Ealy and colleagues 80 identified two other genes within the TGFβ1 pathway that are significantly expressed in both groups: PF4, which selectively prevents TGFβ1 from binding to its type I receptor and may inhibit bone resorption if overexpressed by downregulating TGFβ1 signalling, and IBSP, which is influenced by TGFβ1 signalling in rats and has been found to be downregulated in otosclerosis, along with TGFβ1.
Bone morphogenic proteins
BMP2 and BMP4 also belong to the TGFβ1 signalling network and are crucial in various molecular processes, including bone homeostasis 81. A study by Schrauwen 82 identified a correlation between otosclerosis and specific SNPs: rs3178250T > C in the 3’ UTR of BMP2 and rs17563, p.(Val152Ala) in BMP4. These SNPs were analysed in an Indian population, with only the BMP4 SNP showing a significant association 64. However, when these SNPs were examined in Tunisian and Hungarian populations, no association was found, likely due to insufficient study power 66,74. Further research by Ealy et al. 83 in a German cohort found no link between common variants in BMP2 and BMP4 and otosclerosis, although 4 rare variants – including 2 missense mutations, one large deletion, and one synonymous variant – were exclusively found in affected individuals. Functional assays revealed that the large deletion in BMP2 and the missense mutation p.(Asn150Lys) in BMP4 led to decreased Smad receptor phosphorylation 83. Additional studies in an Indian cohort demonstrated elevated levels of BMP2 and BMP4 in otosclerotic stapes tissues, reinforcing the involvement of these proteins in otosclerosis 64.
TNFRSF11B
The gene TNFRSF11B is responsible for coding OPG, which acts as a decoy receptor for the RANKL. Research has highlighted OPG’s involvement in otosclerosis, with studies showing reduced OPG mRNA expression in stapes tissue from patients compared to normal tissue 8.
Alterations in the expression of OPG have been associated with diseases such as osteopetrosis 26, osteoporosis 27, otosclerosis 19,28, juvenile and adult Paget’s disease of bone 29, and celiac disease characterised by high-turnover osteoporosis 30.
In genetic studies focusing on otosclerosis, a SNP in TNFRSF11B, rs1485286, displayed marginal significance in a Belgian-Dutch male population (p value 0.049) 82. Meanwhile, analysis of 12 Italian patients from otosclerosis-affected families did not reveal pathological mutations; however, the polymorphism rs2073618 was present in 10 of these patients. Further sequencing of the polymorphism in 98 unrelated patients did not show an association with otosclerosis 84. However, in an Indian male population, SNP rs2073618 was significantly linked to otosclerosis 8. Meta-analyses incorporating data from Italian and Indian populations affirmed the association of this SNP with the condition 8. A subsequent meta-analysis involving Tunisian, Indian, and Italian samples also supported this association 85.
Role of inflammation and oxidative stress
The molecular profile of the otic capsule is markedly different from other bones in the body, characterised by a down-regulation of proinflammatory cytokines, and an up-regulation of anti-inflammatory cytokines 18, to the point that proinflammatory cytokines in the cochlea are generally considered markers of disease and have been associated with HL, with anatomical communication between the otic capsule and the cochlea perilymph 19, and with pathological new bone formation within the membranous labyrinth (labyrinthitis ossificans) 35. TNFα and its receptor were reported to be over-expressed during active otosclerosis 86, and because TNFα promotes bone resorption, it may act as a potential catalyst for the dysregulation of bone metabolism in otosclerosis, and may also be a potential contributor to the development of SNHL in otosclerosis 87. Moreover, angiotensin II is a key regulator element for the production of proinflammatory cytokines, it has been reported to be a key factor for inflammation and bone remodelling in otosclerosis, providing a link between the inflammatory and the endocrine pathways in the context of the disease 88.
Oxidative stress has the potential of impacting several cell signalling pathways, and it has been linked to other forms of HL. In otosclerotic patients, immunohistochemical studies have demonstrated an increase in 4-hydroxynonenal (HNE)-protein adducts in comparison with controls. 4-HNE protein adducts are a major bioactive marker of lipid peroxidation which act also as second messengers of free radicals. Although 4-HNE protein adducts were also present in control samples, the primary difference lies in their distribution: they are confined to the periosteal region in controls, whereas in otosclerotic samples HNE-product positive areas are multifocal and irregular 89.
Role of the immune system
It has been suggested that the immune system, particularly an autoimmune reaction targeting the otic capsule, might play a significant role in the development of otosclerosis, and is supported by the fact that immune cells and immune-regulatory factors were discovered in the regions impacted by otosclerosis 87. Initial studies speculated that an autoimmune reaction against type II and other less prevalent collagens could be a potential trigger for the disease 90. The COL2A1 gene, which codes for type II collagen, was targeted for study because this type is plentiful in the globuli interossei, and it has been linked to localised chondrodysplastic lesions 91. Nonetheless, further research involving genetic studies, histological examinations, and immunohistochemical tests have failed to confirm the hypothesis that an autoimmune reaction to collagen is the leading cause of otosclerosis 92.
Additionally, it has been postulated that otosclerosis may be significantly influenced by an autoimmune response initiated by persistent infection with measles virus 87, though definitive proof is still pending.
HLA is an essential part of the human major histocompatibility complex, crucial for presenting antigenic peptides to T cells and controlling the immune response. HLA has been linked to various diseases with an immunologic basis. Although some studies have found associations between certain HLA antigens and otosclerosis, these associations have not been consistent across various studies 87. Nevertheless, the evidence suggesting a significant relationship between HLA and otosclerosis points to an immunological component in the disease, with certain HLA markers potentially affecting susceptibility within specific populations 87. Further investigation is needed to confirm any substantial connections between HLA antigens and otosclerosis.
Role of the endocrine system
Oestrogen
Numerous studies have explored the influence of the endocrine system on the development of otosclerosis, particularly given its more frequent occurrence in females and reports of its manifestation or progression during pregnancy. However, the link with pregnancy is still subject to debate. For instance, a retrospective study did not establish a correlation between the number of pregnancies or children and the progression of otosclerosis-induced HL 93. Proposed mechanisms include the possibility that variants of the estrogen receptor might mediate abnormal bone remodelling in response to oestrogen. Furthermore, oestrogen promotes hyperprolactinaemia, which has been linked to increased bone resorption. This process could counteract the effects of oestrogen itself on bone by diminishing the osteoclast response to RANKL, thereby reducing bone resorption 94.
Renin-angiotensin-aldosterone (RAA) system
The RAA system regulates blood pressure, but is also involved in bone resorption and formation, and specifically angiotensin II has been implicated in key events of inflammation and bone remodelling by its interaction with various growth factors and cytokines 88,89. The hypothesis that the RAA system plays a role in the development of otosclerosis may have been influenced, to some extent, by the observed activation of this pathway during pregnancy 95, coupled with the notion that otosclerosis often appears during or soon after pregnancy. In a 2008 candidate gene-based association study on a French population, polymorphisms in the angiotensin and angiotensinogen-converting enzyme genes associated with higher plasma concentrations of angiotensin II were associated with an increased relative risk of developing otosclerosis 55. Moreover, the same study reported that angiotensin II enhances the secretion of interleukin 6 (IL6) and reduces alkaline phosphatase activity in vitro exclusively in otosclerotic cells, indicating that angiotensin II may play a part in disrupting bone remodelling processes, contributing to the onset of otosclerosis 55. However, these results were not replicated in another candidate gene-based study in a large Belgian-Dutch population 96, and in a study on a Hungarian population 66.
Parathyroid hormone (PTH)
PTH is secreted by the parathyroid glands in response to decreased blood calcium levels, stimulating osteoblasts to release RANKL, which in turn increases bone resorption and liberates more free calcium into the blood 94. Given the significant role that PTH plays in bone metabolism, its involvement in the development of otosclerosis has been suggested. Research has shown that higher concentrations of PTH are necessary to enhance adenylate cyclase activity 97. Additionally, in otosclerotic stapes cell cultures, there is a decreased expression of PTH-PTH-related peptide receptor mRNA accompanied by a reduced cyclic AMP response 98. These findings suggest that a dysfunctional response to PTH may contribute to the abnormal bone turnover observed in otosclerosis.
Vitamin D receptor
Vitamin D stimulates intestinal absorption of calcium, which is associated through an increase in calcitonin, with a decrease in bone resorption 99. Due to their role in bone metabolism, vitamin D and its receptor were proposed as contributing factors to the development of otosclerosis. Yildirim and colleagues 100 genotyped four polymorphisms of the vitamin D receptor gene in a small Turkish population, and found that 3 (Bsm I-rs1544410, Apa I-rs7975232, and Taq I-rs731236) were associated with otosclerosis. However, these results have not yet been replicated in a larger cohort, and therefore it is not possible to draw conclusions on the possible role of vitamin D receptor in the pathophysiology of otosclerosis.
Measles virus
Over the past 30 years, many authors investigated the potential role of measles virus in the development of otosclerosis. The first account of this theory was published in 1986 by McKenna and colleagues 101 who identified filamentous structures resembling measles virus nucleocapsid in osteoblast-like cells in otospongiotic tissue specimens. Over the years, several techniques have been used to investigate this association, including electron microscopy, immunohistochemistry, perilymph analysis, reverse transcription polymerase chain reaction (RTPCR), reverse transcription-quantitative polymerase chain reaction (RTQPCR), and glyceraldehyde 3-phosphate (GADP) to detect mRNA of measles virus in otosclerotic stapes and control samples 102-105. However, other studies could not find evidence of measles virus or of a reaction to it in otosclerotic samples 106,107. The organotropism demonstrated by the measles virus for the otic capsule in humans and primates is due to the complementary cell surface structures CD46 and CD150, which act as virus receptors 108,109. Some studies showed the existence of novel splice variants of CD46 present exclusively in otosclerotic stapes footplates 110-112. An unresolved question lies in the temporality of this relationship: does measles virus trigger the production of new CD46 splicing variants, or do pre-existing unique isoforms enhance virus affinity and facilitate virus replication? The solution might be found in the activity of various regulatory proteins for alternative splicing, which result in distinct expression patterns and modified functions of CD46. In a study by Schrauwen and colleagues 82, 2 (rs2796267 and rs2796270) out of 7 SNPs in CD46 were significantly associated with otosclerosis in males of Belgian-Dutch origin, but the results were not replicated in a French population analysed in the same study. However, the exact role of measles virus in the pathogenesis of otosclerosis and the contribution of genetic factors to this process remain to be described.
Genome wide association studies
One of the limitations of association studies lies in the fact that candidate genes to be tested must be selected in advance based on their potential role in the pathophysiology of otosclerosis. Different from association studies, GWAS is free from the need of hypotheses formulated in advance, and can therefore identify genes associated with the disease that had not been previously considered. GWAS aim to identify association between genetic variants and specific phenotypes (in this case, clinical otosclerosis) by surveying the genome of individuals affected by the disease and controls, and looking for genomic variants that occur more frequently in cases than in controls.
The first GWAS was published in 2009 by Schrauwen and colleagues 113, performed in a population of 1,149 Belgian, Dutch, and French patients and 1,174 matched controls, and identified two regions on chr7q22.1 and chr11q13.1 associated with otosclerosis. The chr7q22.1 region is located near the RELN gene and harbours an intronic SNP rs3914132 found to be strongly associated with the disease. In the same study, a notable correlation with otosclerosis was identified for the SNP rs670358 located on chromosome 11q13.1, a finding that was later confirmed in two separate subpopulations. Additionally, another SNP in this region, rs494252, showed a significant link to otosclerosis in a work by a Tunisian group 114. This SNP is positioned intronically within the CDC42BPG gene and close to EHD1 and MEN1 genes. Both EHD1 and MEN1 are known to be important in the development of bone and cartilage 115,116. However, no subsequent research has definitively shown that these genes play a role in the pathogenesis of otosclerosis.
In 2023, Rämö and colleagues 67 published the largest meta-analysis of GWAS on otosclerosis, utilising data from 3 population-based biobanks that comprised 3,504 cases and 861,198 controls. This study identified 27 risk loci, 23 of which were new, and confirmed associations with otosclerosis for the RELN gene and 3 previously reported candidate genes or linkage regions: TGFβ1, MEPE, and OTSC7. Most of the loci identified were situated near protein-coding genes that are implicated in bone remodelling and mineralisation, which are already associated with severe skeletal disorders such as diaphyseal dysplasia (TGFβ1) and osteopetrosis (CLCN7, TNFSF11).
RELN
The region chr7q22.1, which includes an intronic SNP rs3914132 within the RELN gene, has been consistently identified as having a strong link with otosclerosis, from the first GWAS 113 to subsequent studies across diverse populations 114,117-120. However, some studies lacked sufficient power to reliably detect this association 66,119. Priyadarshi et al. 117 conducted a meta-analysis using a cumulative population from multiple studies, comprising 2,670 cases and 2,812 controls, and reported a significant association of the rs3914132 polymorphism with otosclerosis across different populations and genetic models.
The RELN gene, responsible for producing the reelin protein, plays a crucial role in neural migration and positioning in the developing brain. Reelin is produced exclusively by neural tissues and is implicated in the development of several neurological disorders 121. Disruptions in reelin signalling have been reported in diseases such as bipolar disorder, schizophrenia, and autism 122-124.
The role of reelin in bone metabolism remains largely unclear. However, research by Dou and colleagues 125 on multiple myeloma suggests that reelin significantly influences bone formation and the balance between osteolysis and osteogenesis. High levels of reelin are expressed by osteocytes, the bone’s mechanosensing cells 126, and the protein may contribute to the mechanosensory adaptation mechanism of bone remodelling, as it is detected with elevated expression in limbs compared to skull bones 127. Moreover, distinct expressions of reelin have been observed in the stapes tissues of humans with otosclerosis 120, and a recent study demonstrated the role of RELN variation in familial ankylosing spondylitis, further strengthening the role of this gene in disorders of bone remodelling 128. However, the exact mechanism of RELN in the pathogenesis of otosclerosis remains unclear.
Next generation sequencing
By massive parallel DNA sequencing (sequencing millions of fragments simultaneously per run), NGS allows a high-throughput sequencing of the complete genome, the exome (meaning all coding regions), or selected custom-panels of genes. By being relatively fast and cost-effective, this approach has broken down the limitations associated with sequencing only targeted genes, and has allowed the identification of new genes involved in the pathogenesis of otosclerosis: SERPINF1, ACAN, and MEPE.
SERPINF1
In 2016, Ziff and colleagues 129 identified multiple missense mutations in the serpin peptidase inhibitor-clade F (SERPINF1) gene using various genetic techniques, including whole exome sequencing (WES), on 4 families that exhibited an autosomal dominant inheritance pattern of otosclerosis. SERPINF1 encodes pigment epithelium-derived growth factor (PEDF), a potent angiogenesis inhibitor and a known regulator of bone remodelling. Angiogenesis is a key feature of otosclerosis, and is associated with both Schwartze’s sign and the increased promontory blood flow observed in Doppler flowmetry 130. Moreover, mutations in SERPINF1 are linked to the rare type 4 osteogenesis imperfecta, another bone remodelling disorder 131. However, a larger study conducted in 2019 by Valgaeren et al. 132 on 1,604 unrelated patients, 62 probands from large families, and 1,538 controls, only found 3 missense variants previously reported by Ziff et al. 129 (c.167C > G, c.331G > A, c.392C > A) in 5 patients and 4 controls. Familial analysis identified 12 variants in all affected family members; however, these variants were also frequently found in the control population, which complicates establishing a pathogenic role. None of the variants reported by Ziff and colleagues 129 were found in any of the 62 large families studied. Additionally, a study by Richard et al. 78 in 2015 using proteomic analysis and immunostaining found decreased expression of SERPINF1-012 in otosclerosis patients, with or without SERPINF1 mutations; however, these results have not been replicated.
ACAN
The ACAN gene, located at the OTSC1 locus, encodes aggrecan, the primary non-collagenous component of the cartilaginous extracellular matrix, which is essential for cartilage function and skeletal development 38. The potential role of ACAN in otosclerosis was first suggested by Dawson in 2018 at the Molecular Biology of Hearing and Deafness meeting. He reported the findings from 19 probands from large otosclerosis families who were tested using WES, followed by targeted NGS of 61 candidate genes in 160 familial otosclerosis patients. Notably, more than 20% of these patients carried rare variants of the ACAN gene. In a subsequent study in 2021, Højland and colleagues 133 sequenced the entire ACAN gene – including all coding regions, exon-intron boundaries, and untranslated regions (UTRs) – in 1,468 unrelated patients, 29 familial cases, and 1,437 unscreened controls. This study identified 14 nonsense and missense variants. ACAN is distinguished by a remarkable spectrum of variants in terms of number, effect size, allele frequency, and direction of effect. Specifically, some variants showed strong effects but low frequency, resembling the transmission pattern of monogenic diseases, while others had minimal effect sizes and were more common, serving as susceptibility factors.
MEPE
The MEPE gene encodes a matrix extracellular phosphoglycoprotein with a role in bone homeostasis, suppression of renal calcification, and regulation of serum phosphate. In a study on a large Turkish family affected by hereditary congenital facial paresis and a mixed form of HL similar to otosclerosis, a mutation of the MEPE gene segregating with the phenotype was reported 134. To confirm this finding, a large case-control study was performed, including 123 members from 62 families, 1,604 cases, and 1,538 controls. This study identified 6 heterozygous frameshift and nonsense variants in 19 patients and 3 unscreened controls, indicating a relatively low frequency of these variants in the population but a high effect size. These results were replicated in the large GWAS meta-analysis by Rämö et al. 67, which also reported the dynamic changes in MEPE expression throughout postnatal development in the murine inner ear by immunostaining, going from a more diffused expression at 2 days of life to a limited expression to mature osteocytes at 3 months of age.
Conclusions
Overall, the hereditary pattern in otosclerosis is predominantly complex, involving both environmental and genetic factors, a model already described for other relatively common diseases such as age-related HL and coronary artery and Alzheimer’s disease. Unlike other genetic disorders, where linkage analysis, positional cloning, and GWAS have led to the identification of many causative genes, the genetics of otosclerosis remain largely unidentified. Linkage analysis of monogenic forms of otosclerosis has led to the identification of 9 loci, but the genes responsible and their variants have yet to be extensively described. So far, the most promising results have come from GWAS, which identified strong associations with novel candidate regions. The use of NGS to zoom into these candidate genes in search of causative variants has recently led to the identification of large-effect risk factors in MEPE 134 and variants with high variation in frequency and effect size in ACAN 133, highlighting the potential of this approach.
Apart from genetic studies, epigenetic analyses could provide valuable insights into the pathophysiology of otosclerosis by correlating the impact of environmental factors on local gene expression, as has been described in numerous other complex diseases. A limitation of this approach is the need to use samples of stapes tissue, in contrast with genetic studies that mainly use DNA drawn from blood samples. Additionally, future studies should include clinical data, especially audiometric testing and temporal bone imaging, for better correlation with genetics and pathophysiology, as most large databases have not included such information. An integrated approach, utilising various genetic and epigenetic techniques in conjunction with clinical data and possibly aided by new bioinformatic techniques, will likely provide a better understanding of the pathophysiology of otosclerosis.
Conflict of interest statement
The authors declare no conflict of interest.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contributions
SC, FL, LB: conceived the initial idea for the narrative review and developed the structure of the article; GF, FF: performed the literature review and critically analyzed the sources. All authors contributed significantly to drafting the manuscript, revising it for important intellectual content, and approving the final version to be submitted. Each author has read and agreed to the published version of the manuscript.
Ethical consideration
Not applicable.
History
Received: September 20, 2024
Accepted: April 7, 2025
Figures and tables
| Locus | Position | Study | Investigated candidate genes | Family countries of origin |
|---|---|---|---|---|
| OTSC 1 | 15q25-26 (14.5 Mb) | Tomek et al., 1998 | / | Southern India, Tunisia |
| OTSC 2 | 7q34-36 (16 Mb) | Schrauwen et al., 2010; Van Den Bogaert et al., 2001 | TRB locus, ATP6V0A4, CLEC5A, EPHA1, EPHB6, HIPK2, KLRG2, LUC7L2, MKRN1, PIP, PRSS2, SSBP1, TRIM24, TRPV5, TRPV6 | Belgium, England |
| OTSC 3 | 6p21.3-22.3 (17.4 Mb) | Ali et al., 2007; Chen et al., 2002 | / | Cyprus, Tunisia |
| OTSC 4 | 16q21-23.2 (10 Mb) | Brownstein et al., 2006 | / | Israel |
| OTSC 5 | 3q22-24 (15.5 Mb) | Van Den Bogaert et al., 2004 | PCOLCE2, CHST2 | The Netherlands |
| OTSC 7 | 6q22.3-6q23.3 (16.5 Mb) | Thys et al., 2007 | COL12A1, COL9A1 | Greece, The Netherlands |
| OTSC 8 | 9p13.1-q21.11 (34.16 Mb) | Bel Hadj Ali et al., 2008 | TJP2, TRPM3, KLF9 | Tunisia |
| OTSC 10 | 1q41-44 (26.1 Mb | Schrauwen et al., 2011 | TGFB2, AGT | The Netherlands |
| OTSC 11 | 16q24.1 (9.96 Mb) | Abdelfatah et al., 2022 | FOXL1 | Canada |
References
- Schrauwen I, Van Camp G. The etiology of otosclerosis: a combination of genes and environment. Laryngoscope 2010;120:1195-1202. https://doi.org/10.1002/lary.20934
- Toynbee J. Pathological and surgical observations on the diseases of the ear. Med Chir Trans 1861;24:190-205. https://doi.org/10.1177/095952874102400115
- Morrison AW. Genetic factors in otosclerosis. Ann R Coll Surg Engl 1967;41:202-237.
- Hernandez-Orozco F, Courtney GT. Genetic aspects of clinical otosclerosis. Ann Otol Rhinol Laryngol 1964;73:632-644. https://doi.org/10.1177/000348946407300306
- Rudic M, Keogh I, Wagner R, et al. The pathophysiology of otosclerosis: review of current research. Hear Res 2015;330:51-56. https://doi.org/10.1016/j.heares.2015.07.014
- Altmann F, Glasgold A, Macduff JP. The incidence of otosclerosis as related to race and sex. Ann Otol Rhinol Laryngol 1967;76:377-392. https://doi.org/10.1177/000348946707600207
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature 2003;423:337-342. https://https://doi.org/10.1038/nature01658
- Priyadarshi S, Ray CS, Biswal NC, et al. Genetic association and altered gene expression of osteoprotegerin in otosclerosis patients. Ann Hum Genet 2015;79:225-237. https://doi.org/10.1111/ahg.12118
- Ney JT, Juhasz-Boess I, Gruenhage F, et al. Genetic polymorphism of the OPG gene associated with breast cancer. BMC Cancer 2013;13:40. https://doi.org/10.1186/1471-2407-13-40
- Cundy T, Hegde M, Naot D, et al. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet 2002;11:2119-2127. https://doi.org/10.1093/hmg/11.18.2119
- Anson BJ, Bast TH. The development of the otic capsule in the region of surgical fenestration. Ann Otol Rhinol Laryngol 1949;58:739-750. https://doi.org/10.1177/000348944905800311
- Sørensen MS, Jørgensen MB, Bretlau P. Distribution of bone remodeling units in the otic capsule of the rabbit. A semiquantitative morphometric study. Acta Otolaryngol 1992;112:462-469. https://doi.org/10.3109/00016489209137427
- Frisch T, Sørensen MS, Overgaard S, et al. Volume-referent bone turnover estimated from the interlabel area fraction after sequential labeling. Bone 1998;22:677-682. https://doi.org/10.1016/s8756-3282(98)00050-7
- Sørensen MS, Bretlau P, Jørgensen MB. Quantum type bone remodeling in the otic capsule of the pig. Acta Otolaryngol 1990;110:217-223. https://doi.org/10.3109/00016489009122540
- Anson BJ, Bast TH, Cauldwell EW. The development of the auditory ossicles, the otic capsule and the extracapsular tissues. Ann Otol Rhinol Laryngol 1948;57:603-632. https://doi.org/10.1177/000348944805700303
- Frisch T, Sørensen MS, Overgaard S, et al. Estimation of volume referent bone turnover in the otic capsule after sequential point labeling. Ann Otol Rhinol Laryngol 2000;109:33-39. https://doi.org/10.1177/000348940010900106
- Sørensen MS, Jørgensen MB, Bretlau P. Remodeling patterns in the bony otic capsule of the dog. Ann Otol Rhinol Laryngol 1991;100:751-758. https://doi.org/10.1177/000348949110000913
- Stankovic KM, Adachi O, Tsuji K, et al. Differences in gene expression between the otic capsule and other bones. Hear Res 2010;265:83-89. https://doi.org/10.1016/j.heares.2010.02.006
- Zehnder AF, Kristiansen AG, Adams JC, et al. Osteoprotegerin in the inner ear may inhibit bone remodeling in the otic capsule. Laryngoscope 2005;115:172-177. https://doi.org/10.1097/01.mlg.0000150702.28451.35
- Cawley KM, Bustamante-Gomez NC, Guha AG, et al. Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep 2020;32:108052. https://doi.org/10.1016/j.celrep.2020.108052
- Zehnder AF, Kristiansen AG, Adams JC, et al. Osteoprotegrin knockout mice demonstrate abnormal remodeling of the otic capsule and progressive hearing loss. Laryngoscope 2006;116:201-206. https://doi.org/10.1097/01.mlg.0000191466.09210.9a
- Nielsen MC, Martin-Bertelsen T, Friis M, et al. Differential gene expression in the otic capsule and the middle ear – An annotation of bone-related signaling genes. Otol Neurotol 2015;36:727-732. https://doi.org/10.1097/MAO.0000000000000664
- Kao SY, Katsumi S, Han D, et al. Postnatal expression and possible function of RANK and RANKL in the murine inner ear. Bone 2021;145:115837. https://doi.org/10.1016/j.bone.2020.115837
- Lee Y, Kim HJ, Park CK, et al. Novel extraneural role of neurite outgrowth inhibitor A: modulation of osteoclastogenesis via positive feedback regulation of nuclear factor of activated T cell cytoplasmic 1. J Bone Miner Res 2012;27:1043-1054. https://doi.org/10.1002/jbmr.1561
- Kao SY, Kempfle JS, Jensen JB, et al. Loss of osteoprotegerin expression in the inner ear causes degeneration of the cochlear nerve and sensorineural hearing loss. Neurobiol Dis 2013;56:25-33. https://doi.org/10.1016/j.nbd.2013.04.008.
- Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 1997;89:309-319. https://doi.org/10.1016/s0092-8674(00)80209-3
- Mizuno A, Amizuka N, Irie K, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun 1998;247:610-615. https://doi.org/10.1006/bbrc.1998.8697
- Karosi T, Csomor P, Szalmás A, et al. Osteoprotegerin expression and sensitivity in otosclerosis with different histological activity. Eur Arch Otorhinolaryngol 2011;268:357-365. https://doi.org/10.1007/s00405-010-1404-y
- Daroszewska A, Hocking LJ, McGuigan FE, et al. Susceptibility to Paget’s disease of bone is influenced by a common polymorphic variant of osteoprotegerin. J Bone Miner Res 2004;19:1506-1511. https://doi.org/10.1359/JBMR.040602
- Riches PL, McRorie E, Fraser WD, et al. Osteoporosis associated with neutralizing autoantibodies against osteoprotegerin. N Engl J Med 2009;361:1459-1465. https://doi.org/10.1056/NEJMoa0810925
- Chang W, ten Dijke P, Wu DK. BMP pathways are involved in otic capsule formation and epithelial-mesenchymal signaling in the developing chicken inner ear. Dev Biol 2002;251:380-394. https://doi.org/10.1006/dbio.2002.0822
- Gerlach LM, Hutson MR, Germiller JA, et al. Addition of the BMP4 antagonist, noggin, disrupts avian inner ear development. Development 2000;127:45-54. https://doi.org/10.1242/dev.127.1.45
- Daluiski A, Engstrand T, Bahamonde ME, et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet 2001;27:84-88. https://doi.org/10.1038/83810
- Gamer LW, Cox K, Carlo JM, et al. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev Dyn 2009;238:2374-2381. https://doi.org/10.1002/dvdy.22048
- Hartnick CJ, Kim HH, Chute PM, et al. Preventing labyrinthitis ossificans: the role of steroids. Arch Otolaryngol Head Neck Surg 2001;127:180-183. https://doi.org/10.1001/archotol.127.2.180
- Schrauwen I, Venken K, Vanderstraeten K, et al. Involvement of T-cell receptor-beta alterations in the development of otosclerosis linked to OTSC2. Genes Immun 2010;11:246-253. https://doi.org/10.1038/gene.2010.3
- Abdelfatah N, Mostafa AA, French CR, et al. A pathogenic deletion in Forkhead Box L1 (FOXL1) identifies the first otosclerosis (OTSC) gene. Hum Genet 2022;141:965-979. https://doi.org/10.1007/s00439-021-02381-1
- Tomek MS, Brown MR, Mani SR, et al. Localization of a gene for otosclerosis to chromosome 15q25-q26. Hum Mol Genet 1998;7:285-290. https://doi.org/10.1093/hmg/7.2.285
- Declau F, Van Spaendonck M, Timmermans JP, et al. Prevalence of otosclerosis in an unselected series of temporal bones. Otol Neurotol 2001;22:596-602. https://doi.org/10.1097/00129492-200109000-00006
- McKenna MJ, Kristiansen AG, Tropitzsch AS. Similar COL1A1 expression in fibroblasts from some patients with clinical otosclerosis and those with type I osteogenesis imperfecta. Ann Otol Rhinol Laryngol 2002;111:184-189. https://doi.org/10.1177/000348940211100214
- Van Den Bogaert K, Govaerts PJ, Schatteman I, et al. A second gene for otosclerosis, OTSC2, maps to chromosome 7q34-36. Am J Hum Genet 2001;68:495-500. https://doi.org/10.1086/318185
- Frenz DA, Liu W. Effect of retinoic acid on otic capsule chondrogenesis in high-density culture suggests disruption of epithelial-mesenchymal interactions. Teratology 1997;56:233-240. https://doi.org/10.1002/(SICI)1096-9926(199710)56:4<233::AID-TERA1>3.0.CO;2-#
- Cai L, Xu Y, Liu Y, et al. Study on genetic factors of otosclerosis: review. Ann Otolaryngol Rhinol 2022;9:1289.
- Chen W, Campbell CA, Green GE, et al. Linkage of otosclerosis to a third locus (OTSC3) on human chromosome 6p21.3-22.3. J Med Genet 2002;39:473-477. https://doi.org/10.1136/jmg.39.7.473
- García E, Marcos-Gutiérrez C, del Mar Lorente M, et al. RYBP, a new repressor protein that interacts with components of the mammalian Polycomb complex, and with the transcription factor YY1. EMBO J 1999;18:3404-3418. https://doi.org/10.1093/emboj/18.12.3404
- Riquet FB, Tan L, Choy BK, et al. YY1 is a positive regulator of transcription of the Col1a1 gene. J Biol Chem 2001;276:38665-38672. https://doi.org/10.1074/jbc.M009881200
- McGuirt WT, Prasad SD, Griffith AJ, et al. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat Genet 1999;23:413-419. https://doi.org/10.1038/70516
- Gregoriadis S, Zervas J, Varletzidis E, et al. HLA antigens and otosclerosis. A possible new genetic factor. Arch Otolaryngol 1982;108:769-771. https://doi.org/10.1001/archotol.1982.00790600013004
- Brownstein Z, Goldfarb A, Levi H, et al. Chromosomal mapping and phenotypic characterization of hereditary otosclerosis linked to the OTSC4 locus. Arch Otolaryngol Head Neck Surg 2006;132:416-424. https://doi.org/10.1001/archotol.132.4.416
- Van Den Bogaert K, De Leenheer EM, Chen W, et al. A fifth locus for otosclerosis, OTSC5, maps to chromosome 3q22-24. J Med Genet 2004;41:450-453. https://doi.org/10.1136/jmg.2004.018671
- Thys M, Van Den Bogaert K, Iliadou V, et al. A seventh locus for otosclerosis, OTSC7, maps to chromosome 6q13-16.1. Eur J Hum Genet 2007;15:362-368. https://doi.org/10.1038/sj.ejhg.5201761
- Bel Hadj Ali I, Thys M, Beltaief N, et al. A new locus for otosclerosis, OTSC8, maps to the pericentromeric region of chromosome 9. Hum Genet 2008;123:267-272. https://doi.org/10.1007/s00439-008-0470-3
- Schrauwen I, Weegerink NJ, Fransen E, et al. A new locus for otosclerosis, OTSC10, maps to chromosome 1q41-44. Clin Genet 2011;79:495-497. https://doi.org/10.1111/j.1399-0004.2010.01576.x
- Thys M, Schrauwen I, Vanderstraeten K, et al. The coding polymorphism T263I in TGF-β1 is associated with otosclerosis in two independent populations. Hum Mol Genet 2007;16:2021-2030. https://doi.org/10.1093/hmg/ddm150
- Imauchi Y, Jeunemaitre X, Boussion M, et al. Relation between renin-angiotensin-aldosterone system and otosclerosis: a genetic association and in vitro study. Otol Neurotol 2008;29:295-301. https://doi.org/10.1097/mao.0b013e318164d12c
- Lam EW, Brosens JJ, Gomes AR, et al. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer 2013;13:482-495. https://doi.org/10.1038/nrc3539.
- Brown DJ, Kim TB, Petty EM, et al. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet 2002;71:618-624. https://doi.org/10.1086/342067
- Tavernier LJM, Fransen E, Valgaeren H, et al. Genetics of otosclerosis: finally catching up with other complex traits? Hum Genet 2022;141:939-950. https://doi.org/10.1007/s00439-021-02357-1
- McKenna MJ, Kristiansen AG, Bartley ML, et al. Association of COL1A1 and otosclerosis: evidence for a shared genetic etiology with mild osteogenesis imperfecta. Am J Otol 1998;19:604-610.
- McKenna MJ, Nguyen-Huynh AT, Kristiansen AG. Association of otosclerosis with Sp1 binding site polymorphism in COL1A1 gene: evidence for a shared genetic etiology with osteoporosis. Otol Neurotol 2004;25:447-450. https://doi.org/10.1097/00129492-200407000-00008
- Zhang Y, Tang Q, Xue R, et al. Analysis of the genetic characteristics of a Chinese family with otosclerosis. Ear Nose Throat J 2021;100:774S-780S. https://doi.org/10.1177/0145561320910627
- Chen W, Meyer NC, McKenna MJ, et al. Single-nucleotide polymorphisms in the COL1A1 regulatory regions are associated with otosclerosis. Clin Genet 2007;71:406-414. https://doi.org/10.1111/j.1399-0004.2007.00794.x
- Schrauwen I, Khalfallah A, Ealy M, et al. COL1A1 association and otosclerosis: a meta-analysis. Am J Med Genet A 2012;158A:1066-1070. https://doi.org/10.1002/ajmg.a.35276
- Hansdah K, Singh N, Bouzid A, et al. Evaluation of the genetic association and mRNA expression of the COL1A1, BMP2, and BMP4 genes in the development of otosclerosis. Genet Test Mol Biomarkers 2020;24:343-351. https://doi.org/10.1089/gtmb.2019.0235
- Rodríguez L, Rodríguez S, Hermida J, et al. Proposed association between the COL1A1 and COL1A2 genes and otosclerosis is not supported by a case-control study in Spain. Am J Med Genet A 2004;128A:19-22. https://doi.org/10.1002/ajmg.a.30074
- Sommen M, Van Camp G, Liktor B, et al. Genetic association analysis in a clinically and histologically confirmed otosclerosis population confirms association with the TGFB1 gene but suggests an association of the RELN gene with a clinically indistinguishable otosclerosis-like phenotype. Otol Neurotol 2014;35:1058-1064. https://doi.org/10.1097/MAO.0000000000000334
- Rämö JT, Kiiskinen T, Seist R, et al. Genome-wide screen of otosclerosis in population biobanks: 27 loci and shared associations with skeletal structure. Nat Commun 2023;14:157. https://doi.org/10.1038/s41467-022-32936-3
- Janssens K, ten Dijke P, Janssens S, et al. Transforming growth factor-beta1 to the bone. Endocr Rev 2005;26:743-774. https://doi.org/10.1210/er.2004-0001
- Bodo M, Venti G, Baroni T, et al. Phenotype of in vitro human otosclerotic cells and its modulation by TGF beta. Cell Mol Biol (Noisy-le-grand) 1995;41:1039-1049.
- Chen G, Deng C, Li YP. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 2012;8:272-288. https://doi.org/10.7150/ijbs.2929
- Ignotz RA, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 1986;261:4337-4345.
- Weber PC, Cunningham CD 3rd, Schulte BA. Potassium recycling pathways in the human cochlea. Laryngoscope 2001;111:1156-1165. https://doi.org/10.1097/00005537-200107000-00006
- Frenz DA, Van de Water TR, Galinovíc-Schwartz V. Transforming growth factor beta: does it direct otic capsule formation? Ann Otol Rhinol Laryngol 1991;100:301-307. https://doi.org/10.1177/000348949110000407
- Khalfallah A, Schrauwen I, Mnejja M, et al. Association of COL1A1 and TGFB1 polymorphisms with otosclerosis in a Tunisian population. Ann Hum Genet 2011;75:598-604. https://doi.org/10.1111/j.1469-1809.2011.00665.x
- Tshifularo M, Joseph CA. Otosclerosis and TGF-beta 1 gene in black South Africans. S Afr Med J 2008;98:720-723.
- Thys M, Schrauwen I, Vanderstraeten K, et al. Detection of rare nonsynonymous variants in TGFB1 in otosclerosis patients. Ann Hum Genet 2009;73:171-175. https://doi.org/10.1111/j.1469-1809.2009.00505.x
- Priyadarshi S, Hansdah K, Ray CS, et al. Otosclerosis associated with a de novo mutation -832G > A in the TGFB1 gene promoter causing a decreased expression level. Sci Rep 2016;6:29572. https://doi.org/10.1038/srep29572
- Richard C, Doherty JK, Fayad JN, et al. Identification of target proteins involved in cochlear otosclerosis. Otol Neurotol 2015;36:923-931. https://doi.org/10.1097/MAO.0000000000000680
- Wu M, Chen G, Li YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 2016;4:16009. https://doi.org/10.1038/boneres.2016.9
- Ealy M, Chen W, Ryu GY, et al. Gene expression analysis of human otosclerotic stapedial footplates. Hear Res 2008;240:80-86. https://doi.org/ 10.1016/j.heares.2008.03.001
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors 2004;22:233-241. https://doi.org/10.1080/08977190412331279890
- Schrauwen I, Thys M, Vanderstraeten K, et al. Association of bone morphogenetic proteins with otosclerosis. J Bone Miner Res 2008;23:507-516. https://doi.org/10.1359/jbmr.071112
- Ealy M, Meyer NC, Corchado JC, et al. Rare variants in BMP2 and BMP4 found in otosclerosis patients reduce Smad signaling. Otol Neurotol 2014;35:395-400. https://doi.org/10.1097/MAO.0000000000000244
- Iossa S, Morello G, Esposito T, et al. Exclusion of TNFRSF11B as candidate gene for otosclerosis in Campania population. Indian J Otolaryngol Head Neck Surg 2014;66:297-301. https://doi.org/10.1007/s12070-014-0706-6
- Bouzid A, Tekari A, Jbeli F, et al. Osteoprotegerin gene polymorphisms and otosclerosis: an additional genetic association study, multilocus interaction and meta-analysis. BMC Med Genet 2020;21:122. https://doi.org/10.1186/s12881-020-01036-8
- Csomor P, Sziklai I, Karosi T. TNF-alpha receptor expression correlates with histologic activity of otosclerosis. Otol Neurotol 2009;30:1131-1137. https://doi.org/10.1097/MAO.0b013e3181be6af4
- Babcock TA, Liu XZ. Otosclerosis: from genetics to molecular biology. Otolaryngol Clin North Am 2018;51:305-318. https://doi.org/10.1016/j.otc.2017.11.002
- Rudic M, Nguyen C, Nguyen Y, et al. Effect of angiotensin II on inflammation pathways in human primary bone cell cultures in otosclerosis. Audiol Neurootol 2012;17:169-178. https://doi.org/10.1159/000335098
- Rudic M, Milković L, Žarković K, et al. The effects of angiotensin II and the oxidative stress mediator 4-hydroxynonenal on human osteoblast-like cell growth: possible relevance to otosclerosis. Free Radic Biol Med 2013;57:22-28. https://doi.org/10.1016/j.freeradbiomed.2012.11.023
- Yoo TJ. Etiopathogenesis of otosclerosis: a hypothesis. Ann Otol Rhinol Laryngol 1984;93:28-33. https://doi.org/10.1177/000348948409300107
- McKenna MJ, Kristiansen AG. The role of measles virus and hereditary in the development of otosclerosis. In: Veldman JE, Passali D, Lim DJ, eds. New frontiers in immunobiology. Monroe (NY): Library Research Associates, Inc.; 2000. pp. 51-56.
- Sølvsten Sørensen M, Nielsen LP, Bretlau P, et al. The role of type II collagen autoimmunity in otosclerosis revisited. Acta Otolaryngol 1988;105:242-247. https://doi.org/10.3109/00016488809097004
- Lippy WH, Berenholz LP, Schuring AG, et al. Does pregnancy affect otosclerosis? Laryngoscope 2005;115:1833-1836. https://doi.org/10.1097/01.MLG.0000187573.99335.85
- Horner KC. The effect of sex hormones on bone metabolism of the otic capsule – An overview. Hear Res 2009;252:56-60. https://doi.org/10.1016/j.heares.2008.12.004
- Schrier RW, Dürr JA. Pregnancy: an overfill or underfill state. Am J Kidney Dis 1987;9:284-289. https://doi.org/10.1016/s0272-6386(87)80123-3
- Schrauwen I, Thys M, Vanderstraeten K, et al. No evidence for association between the renin-angiotensin-aldosterone system and otosclerosis in a large Belgian-Dutch population. Otol Neurotol 2009;30:1079-1083. https://doi.org/10.1097/MAO.0b013e3181ab3058
- Fanó G, Venti-Donti G, Belia S, et al. PTH induces modification of transductive events in otosclerotic bone cell cultures. Cell Biochem Funct 1993;11:257-261. https://doi.org/10.1002/cbf.290110406
- Grayeli AB, Sterkers O, Roulleau P, et al. Parathyroid hormone-parathyroid hormone-related peptide receptor expression and function in otosclerosis. Am J Physiol 1999;277. https://doi.org/10.1152/ajpendo.1999.277.6.E1005
- Carmeliet G, Dermauw V, Bouillon R. Vitamin D signaling in calcium and bone homeostasis: a delicate balance. Best Pract Res Clin Endocrinol Metab 2015;29:621-631. https://doi.org/10.1016/j.beem.2015.06.001
- Yıldırım YS, Apuhan T, Düzenli S, et al. Otosclerosis and vitamin D receptor gene polymorphism. Am J Otolaryngol 2013;34:454-457. https://doi.org/10.1016/j.amjoto.2013.03.016
- McKenna MJ, Mills BG, Galey FR, et al. Filamentous structures morphologically similar to viral nucleocapsids in otosclerotic lesions in two patients. Am J Otol 1986;7:25-28.
- Potocka-Bakłażec M, Sakowicz-Burkiewicz M, Kuczkowski J, et al. Expression of TNF-α, OPG, IL-1β and the presence of the measles virus RNA in the stapes of the patients with otosclerosis. Eur Arch Otorhinolaryngol 2015;272:1907-1912. https://doi.org/10.1007/s00405-014-3008-4
- Karosi T, Kónya J, Szabó LZ, et al. Measles virus prevalence in otosclerotic foci. Adv Otorhinolaryngol 2007;65:93-106. https://doi.org/10.1159/000098677
- Niedermeyer HP, Gantumur T, Neubert WJ, et al. Measles virus and otosclerosis. Adv Otorhinolaryngol 2007;65:86-92. https://doi.org/10.1159/000098676
- McKenna MJ, Kristiansen AG, Haines J. Polymerase chain reaction amplification of a measles virus sequence from human temporal bone sections with active otosclerosis. Am J Otol 1996;17:827-830.
- Komune N, Ohashi M, Matsumoto N, et al. No evidence for an association between persistent measles virus infection and otosclerosis among patients with otosclerosis in Japan. J Clin Microbiol 2012;50:626-632. https://doi.org/10.1128/JCM.06163-11
- Grayeli AB, Palmer P, Tran Ba Huy P, et al. No evidence of measles virus in stapes samples from patients with otosclerosis. J Clin Microbiol 2000;38:2655-2660. https://doi.org/10.1128/JCM.38.7.2655-2660.2000
- Dörig RE, Marcil A, Chopra A, et al. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993;75:295-305. https://doi.org/10.1016/0092-8674(93)80071-l
- Tatsuo H, Ono N, Tanaka K, et al. SLAM (CDw150) is a cellular receptor for measles virus. Nature 2000;406:893-897. https://doi.org/10.1038/35022579
- Karosi T, Szalmás A, Csomor P, et al. Disease-associated novel CD46 splicing variants and pathologic bone remodeling in otosclerosis. Laryngoscope 2008;118:1669-1676. https://doi.org/10.1097/MLG.0b013e31817c133d
- Liktor B, Csomor P, Karosi T. Detection of otosclerosis-specific measles virus receptor (CD46) protein isoforms. ISRN Otolaryngol 2013;2013:479482. https://doi.org/10.1155/2013/479482
- Csomor P, Szalmás A, Kónya J, et al. Restriction analysis of otosclerosis-associated CD46 splicing variants. Eur Arch Otorhinolaryngol 2010;267:219-226. https://doi.org/10.1007/s00405-009-1042-4
- Schrauwen I, Ealy M, Huentelman MJ, et al. A genome-wide analysis identifies genetic variants in the RELN gene associated with otosclerosis. Am J Hum Genet 2009;84:328-338. https://doi.org/10.1016/j.ajhg.2009.01.023
- Khalfallah A, Schrauwen I, Mnaja M, et al. Genetic variants in RELN are associated with otosclerosis in a non-European population from Tunisia. Ann Hum Genet 2010;74:399-405. https://doi.org/10.1111/j.1469-1809.2010.00595.x
- Sowa H, Kaji H, Hendy GN, et al. Menin is required for bone morphogenetic protein 2 and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem 2004;279:40267-40275. https://doi.org/10.1074/jbc.M401312200
- Mintz L, Galperin E, Pasmanik-Chor M, et al. EHD1 – An EH-domain-containing protein with a specific expression pattern. Genomics 1999;59:66-76. https://doi.org/10.1006/geno.1999.5800
- Priyadarshi S, Hansdah K, Singh N, et al. The risks of RELN polymorphisms and its expression in the development of otosclerosis. PLoS One 2022;17:E0269558. https://doi.org/10.1371/journal.pone.0269558
- Schrauwen I, Ealy M, Fransen E, et al. Genetic variants in the RELN gene are associated with otosclerosis in multiple European populations. Hum Genet 2010;127:155-162. https://doi.org/10.1007/s00439-009-0754-2
- Priyadarshi S, Panda KC, Panda AK, et al. Lack of association between SNP rs3914132 of the RELN gene and otosclerosis in India. Genet Mol Res 2010;9:1914-1920. https://doi.org/10.4238/vol9-3gmr890
- Mowat AJ, Crompton M, Ziff JL, et al. Evidence of distinct RELN and TGFB1 genetic associations in familial and non-familial otosclerosis in a British population. Hum Genet 2018;137:357-363. https://doi.org/10.1007/s00439-018-1889-9
- Armstrong NC, Anderson RC, McDermott KW. Reelin: diverse roles in central nervous system development, health and disease. Int J Biochem Cell Biol 2019;112:72-75. https://doi.org/10.1016/j.biocel.2019.04.009
- Fatemi SH, Snow AV, Stary JM, et al. Reelin signaling is impaired in autism. Biol Psychiatry 2005;57:777-787. https://doi.org/10.1016/j.biopsych.2004.12.018
- Goes FS, Willour VL, Zandi PP, et al. Sex-specific association of the Reelin gene with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 2010;153B:549-553. https://doi.org/10.1002/ajmg.b.31018
- Shifman S, Johannesson M, Bronstein M, et al. Genome-wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet 2008;4:E28. https://doi.org/10.1371/journal.pgen.0040028
- Dou A, Zhang Y, Wang Y, et al. Reelin depletion alleviates multiple myeloma bone disease by promoting osteogenesis and inhibiting osteolysis. Cell Death Discov 2021;7:219. https://doi.org/10.1038/s41420-021-00608-8
- Paic F, Igwe JC, Nori R, et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone 2009;45:682-692. https://doi.org/10.1016/j.bone.2009.06.010
- Rawlinson SC, McKay IJ, Ghuman M, et al. Adult rat bones maintain distinct regionalized expression of markers associated with their development. PLoS One 2009;4:E8358. https://doi.org/10.1371/journal.pone.0008358
- Garshasbi M, Mahmoudi M, Razmara E, et al. Identification of RELN variant p.(Ser2486Gly) in an Iranian family with ankylosing spondylitis; the first association of RELN and AS. Eur J Hum Genet 2020;28:754-762. https://doi.org/10.1038/s41431-020-0573-4
- Ziff JL, Crompton M, Powell HR, et al. Mutations and altered expression of SERPINF1 in patients with familial otosclerosis. Hum Mol Genet 2016;25:2393-2403. https://doi.org/10.1093/hmg/ddw106
- Stankovic KM, McKenna MJ. Current research in otosclerosis. Curr Opin Otolaryngol Head Neck Surg 2006;14:347-351. https://doi.org/10.1097/01.moo.0000244194.97301.19
- Wang JY, Liu Y, Song LJ, et al. Novel mutations in SERPINF1 result in rare osteogenesis imperfecta type VI. Calcif Tissue Int 2017;100:55-66. https://doi.org/10.1007/s00223-016-0201-z
- Valgaeren H, Sommen M, Beyens M, et al. Insufficient evidence for a role of SERPINF1 in otosclerosis. Mol Genet Genomics 2019;294:1001-1006. https://doi.org/10.1007/s00438-019-01558-8
- Højland AT, Tavernier LJM, Schrauwen I, et al. A wide range of protective and predisposing variants in aggrecan influence the susceptibility for otosclerosis. Hum Genet 2022;141:951-963. https://doi.org/10.1007/s00439-021-02334-8
- Schrauwen I, Valgaeren H, Tomas-Roca L, et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet Med 2019;21:1199-1208. https://doi.org/10.1038/s41436-018-0300-5
Downloads
License

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Copyright
Copyright (c) 2025 Società Italiana di Otorinolaringoiatria e chirurgia cervico facciale
How to Cite
- Abstract viewed - 2753 times
- PDF downloaded - 471 times